
バイオバーデン/微生物管理関連のオープンセミナーを定期開催しています、詳細はこちらから…。
医療機器QMS省令、ISO13485に関連して滅菌系若しくは微生物管理が要求される製品を取り扱う企業様の場合、定期的に確認が必要となるバイオバーデンという概念があります。
医療機器関連のバイオバーデンは「製品、及び/又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群」として定義されます(ISO 11139:2006)。
一方で製薬分野では「滅菌前の原料及び資料等に製造する微生物の数と種類をいう」(無菌操作法による無菌医薬品の製造に関する指針から)などと定義され、若干のニュアンスの違いが認められます。
医療機器における定義はISOの和訳のため一見掴みづらい印象を持ちますが、意味合いを含め製薬分野の表現を含めて捉えることで少し身近に感じるかもしれません。
バイオバーデンは原材料、資料、最終製品などを対象として実施されます。これに加え、クリーンルーム内の製造環境に存在する微生物菌叢ともリンクを付け考察することで強固なバックデータを保持することが可能になります。
この「環境バイオバーデン」のアプローチは、影響を及ぼす因子を可能な限り抽出して、年間を通した振れ幅を把握を行った上で、原材料、資料、最終製品の菌叢とも相関を取ることが必要になります。
(詳細は環境微生物管理に関するバリデーションを参照 https://www.kea-mgt.com/archives/sem-160906/)
医療機器関連の企業様には微生物専門のセクションを設置しない場合も多く認められます。
一方で製造品目によっては「製品の滅菌性担保」「微生物汚染発生時の究明や考察」などの対応が必要になる場合が生じます。
今回は、医療機器関連の微生物管理の要求事項として「バイオバーデン」の関連情報をまとめてみたいと思います。
ISO 11737-1:2016医療用具の滅菌 微生物学的方法 (バイオバーデンについて)
ISO 11737-1で定義される規格は、JIS T11737-1:2103と相互リンクしており「医療機器、構成部品、原料又は包装上若しくはその内部における生育可能な微生物群の計測および特性付けのための要求事項」と概要説明がついている。
主な要求事項は4項~8項まで。
4項:品質マネジメントシステムに関連する要素
・文書化
・経営者の責任
・製品実現
・不適合製品の管理
が含まれます。
ISO 9001を踏襲した前提条件の位置づけになりますが、バイオバーデンそのものが経営者の責任範囲に含まれ、責任や権限を予め定めることが要求事項になっています。また、文書化の一部には、記録の取扱い内に計算やデータ変換のチェックシステムが含まれることも、9001や13485には出てこないユニークな要求項目です。
5項:製品の選択
・一般
・分割試料
が含まれます。
この項では、選択したサンプルが製造プロセス内の代表であることを根拠を持って説明できることが要求事項になっています。またサンプリングから試験までの時間設定も含まれます。
製品形態として試験をする場合は、想定される汚染がスポット的であるか、全域に渡っているかで部位設定を行わなければなりません。
6項:バイオバーデンの測定及び微生物的特徴づけの方法
・バイオバーデンの測定
→手法選択
→微生物の取り出し
→微生物の培養
→菌数測定
・バイオバーデンの微生物学的特徴づけ(分類同定)
が含まれます。
バイオバーデンを測定するにあたり一番メインとなる項になります。特に測定部では設計と選択手法の妥当性について裏を取ることが要求事項になります。取り出し方法については決定樹を参考にして決めると良いでしょう。
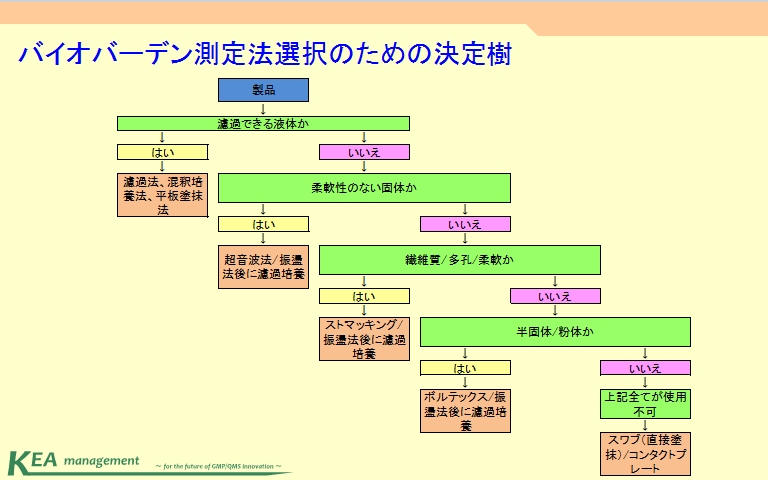
7項:測定方法のバリデーション
・試験方法はバリデートし文書化する
・微生物を取り出す方法の妥当性評価
・補正回数のため回収率設定
・菌数測定の妥当性評価
・分類同定の技術の妥当性評価
が含まれます。
7項では試験系のバリデーションが要求事項として定義され、文書化までが求められます。取り出し方法の妥当性や補正率回収率の設定については、重要な項目になりますので設計段階から組み込むことが必要です。
8項:日常のバイオバーデン測定及びデータの解釈
・サンプルサイズ+頻度を決めたサンプリング計画の文書化
・あらかじめ定めた方法でバイオバーデンの実施
・通常と異なる菌叢の微生物検出時は分離菌株の特性評価を考慮
・滅菌プロセス確立との関連性
・データに基づく許容限界値と逸脱時の処理方法の設定
・一定期間ごとのバイオバーデンデータの傾向分析と許容限界値のレビュー/改定
が含まれます。
初期のバイオバーデン試験の詳細データが不明確な場合、この8項に沿って実施されている事例を多く見受けられます。要求事項では6項、7項をベースとして8項が立案される位置づけです。これにより日常のバイオバーデンそのものがバリデーションされる構造になっています。
現状でサイズやサイクルの根拠が不明確、異常時であるかを検出/確認するための仕組みが決められていない、等の場合は、前項まで戻り設計を強固にしてください。
9項:バイオバーデン測定方法維持
・製品及び製造プロセスの変更時、バイオバーデン変化の可能性をレビュー/記録
・測定方法の変更がある場合、測定結果に与える影響、回収率の面から評価
・初期/再バリデーションのレビューについて定期実施期間を定めて文書化
・再バリデーション結果の記録
が含まれます。
ここでは、日常のバイオバーデン測定が連続的に安定してデータになるための事項が含まれます。。特に再バリデーションの仕組みをシステムとして文書に組み込むことが要求されているため注意が必要です。つまり、初期のバイオバーデン一式のデータをそのまま継続的に適応することが好ましくなく、何らかのタイミングにより定期的な再バリデーションを掛け記録を残すことが必要とされます。
以上、医療機器関連のバイオバーデンの要求事項についてまとめてきました。
2社監査、更新監査時などにこの分野に詳しいオーディターが含まれる場合、指摘や確認を受けることがあります。バイオバーデン試験プロセス自体が様々なバリデーション項目とリンクしています。現手技手法やデータについて根拠ある説明ができない場合は上記要求事項を見直し、堅牢なシステムへと構築することが必要です。
また、バイオバーデン試験及び考察は製品の微生物汚染発生を未然に防ぐことのできる数少ない打ち手です。特に自社資源では対応が難しい専門項目も含まれるため「微生物汚染などの問題が発生」してからの対応ではなく、他のバリデーション項目と足並みをそろえた推進が望まれます。
微生物汚染の原因究明、微生物管理/バイオバーデン試験のお問合せはフォームからお願いいたします
